About
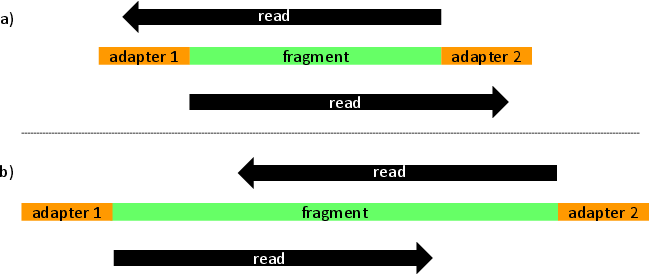
leeHom is a Bayesian maximum a posteriori algorithm for stripping sequencing adapters and merging overlapping portions of reads. Our algorithm is mostly aimed at ancient DNA and Illumina data but can be used for any dataset. As a caveat, leeHom uses heavily quality scores and requires them to be representative of error probabilities. For optimal performance, users can optionally enter a prior that models de probability of observing a specific insert size.
News
- March 3, 2021: leeHom is added to bioconda.
- June 18, 2020: A new maximum-likelihood method for inferring adapter sequences on paired-end data using the --auto parameter.
- August 21, 2019: leeHom can handle UMIs on the ends of insert. If the UMIs were observed twice, a maximum-likelihood inference of the UMI is used.
- March 23, 2018: You can now "trim" bases from the start of the sequencing using "-k N,N" for a single base, "-k NN,NN" for 2 bases, etc.
- May 10, 2016: leeHom is now multi-threaded for both BAM and fastq files. Use "leeHomMulti" with option "-t" for multi-cores. Depending on your I/O reading/writing speeds, the benefits level off after a few cores as I/O is the main bottleneck.
Citing
Our paper describing our algorithm was published in Nucleic Acids Research. To cite us:leeHom: adaptor trimming and merging for Illumina sequencing reads
Gabriel Renaud, Udo Stenzel and Janet Kelso
Nucleic Acids Research 2014 Oct;42(18):e141. doi: 10.1093/nar/gku699.
Documentation
For installation, usage and other topics, please see the README in the software package or use bioconda:
conda install -c bioconda leehom
Support or Contact
Please contact Gabriel Renaud (@grenaud) for further information: